
Chapters IV and V of the MDR 2017 provide detailed guidelines for manufacturers and importers seeking to manufacture or import small quantities of medical devices for specific purposes in India. These licenses enable companies to conduct clinical investigations, testing, evaluation, demonstrations, or training without the need for full manufacturing or import licenses. In this article, we provide an in-depth overview of the test license regulatory process, eligibility requirements, steps involved, necessary documentation, validity, cancellation conditions, and the benefits associated with obtaining a test license.
Eligibility for Medical Test License:
Any importer or manufacturer aiming to import or manufacture small amounts of medical devices for testing, evaluation, demonstration, or training purposes must obtain a test licence approved by the Central Drugs Standard Control Organization (CDSCO). For commercial purposes, a full import or manufacturing licence is required.
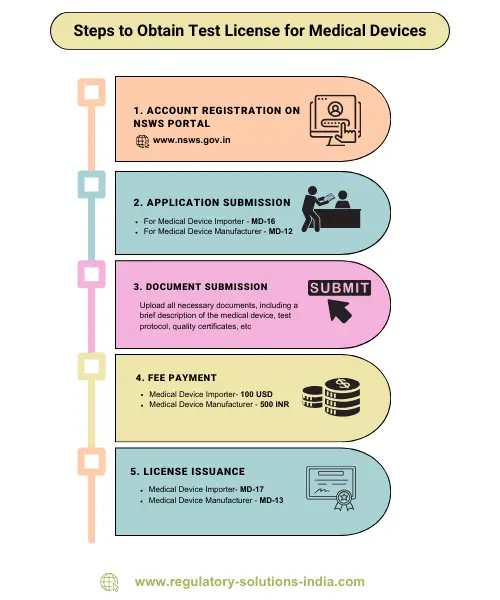
Steps to Apply for Test License:
- Create an account on the NSWS portal and register.
- Apply for approval using Form MD-16 for import or Form MD-12 for manufacture.
- Upload all necessary documents.
- Pay the prescribed fee.
- CDSCO will review the application form and issue a test license in Form MD-17 for import or Form MD-13 for manufacture if found satisfactory.
Documents Required:
- Brief description of the medical device, including intended use, material of construction, and design.
- Justification of the proposed quantity
- Test protocol/Approved clinical investigation plan
- Quality certificates such as QMS
- Labels and Instructions for Use (IFU)
- Undertaking stating the use of the medical device for non-commercial purpose.
- Undertaking from the testing laboratory confirming the provision of required facilities.
Applicable Fees:
1. Test license for manufacture: 500 Rs for each distinct medical device
2. Test license for import: 100USD for each distinct medical device
Validity: The test license remains valid for 3 years from the date of issue.
Cancellation of Test License: If a licensee contravenes any provisions of the rules, the Central Licensing Authority may issue a show cause notice for cancellation. The licensee has the right to appeal to the Central Government within forty-five days from the date of the cancellation order.
Benefits:
1. Facilitates innovation while ensuring compliance with regulatory standards
2. Allows companies to do product testing/ clinical investigation prior to market introduction.
3. Provides opportunity for companies to conduct internal trainings prior to full-scale commercial launch.
Conclusion:
Obtaining a test license for medical devices in India is essential for companies engaging in testing, evaluation, demonstration, or training activities. By adhering to regulatory standards and obtaining expert guidance, companies can navigate the licensing process effectively and contribute to advancing healthcare innovation in India.
Why RSI ?
Regulatory Solutions India (RSI) provides comprehensive regulatory consulting services to assist companies in obtaining test license for medical devices in India. With expertise in eligibility criteria, documentation preparation, submission, and compliance with regulatory requirements, RSI can streamline the licensing process, mitigate risks, and accelerate market entry of your medical device in India.
Contact us for all regulatory needs.
FAQ's
Test License For Manufacturing:
1. What records does the licensee need to maintain under the test license for manufacture?
The licensee must keep a detailed record of the quantity of products manufactured under the test license.
2. How should the medical device be used under the test license for manufacture?
The medical device manufactured under the test license should be used exclusively for clinical investigations, tests, evaluations, examinations, demonstrations, or training at the specified location mentioned in the license.
3. Can Medical Device Officers inspect the manufacturing premises?
Yes, Medical Device Officers have the authority to enter the manufacturing premises, with or without notice, to ensure that only authorised activities are being conducted.
4. What other records are required to be maintained by the licensee?
The licensee should maintain records of the quantity of medical devices manufactured, tested, and stocked, along with their disposition.
Test License For Import:
1. How should the medical device be used under the test license for import?
The medical device imported under the test license should be used exclusively for clinical investigations, tests, evaluations, demonstrations, or training at the specified location mentioned in the license.
2. Can the medical device be taken to places other than those mentioned in the test license?
No, if the medical device needs to be taken to a place other than those specified in the license, written permission must be obtained from the Central Licensing Authority.
What activities should the holder of the test license maintain records of?
The holder of the test license must maintain records of activities undertaken, including the name of the manufacturer, quantity imported, and date of import.
4. What documents should accompany the consignment of medical devices?
The consignment of medical devices should be accompanied by an invoice or statement showing the name and quantity of the medical devices.
5. What should be done with unused medical devices?
Unused medical devices, including in vitro diagnostic medical devices, may be exported or destroyed with intimation to the Central Licensing Authority.



